Autor: Frédéric B. Piel, David J. Weatherall N Engl J Med 2014; 371: 1908-1916. nejm.org DOI: 10.1056/NEJMra1404415. The α-Thalassemias
Introducción
A nivel mundial, las formas más importantes son la α- y la
β-talasemia, que afectan a la producción de las cadenas de α-globina y
β-globina, respectivamente. Aunque la β-talasemia es la forma clínicamente más significativa, la α-talasemia
se produce con una alta frecuencia a lo largo de la zona tropical, casi
llegando a la fijación (un término de la genética de poblaciones que
denota que un alelo mutante de un gen particular se ha convertido en el
único alelo expresado en la población - es decir, que ha llegado a una
frecuencia del 100%) en partes del sur de Asia. Se ha estimado que
alrededor del 5% de la población en todo el mundo porta una variante de
α-talasemia.
Hay evidencia creciente de que la carga sanitaria y económica de las talasemias es cada vez mayor debido al crecimiento de la población, a la transición epidemiológica en regiones tropicales y a las migraciones humanas en otras partes del mundo. (La transición epidemiológica se refiere a un cambio en los patrones de distribución de las edades, la mortalidad, la fecundidad, la esperanza de vida, y las causas de muerte de la población, por lo general reflejado por un cambio de las muertes causadas por enfermedades infecciosas a las muertes causadas por enfermedades crónicas y degenerativas).
El crecimiento de la población conduce a un aumento absoluto del número de nacidos afectados. La transición epidemiológica mejora el diagnóstico de las hemoglobinopatías y la sobrevida de las personas afectadas, aumentando la incidencia de los trastornos. Las migraciones de la población, aunque no siempre conducen a un aumento de la prevalencia global, contribuyen a la propagación de las hemoglobinopatías aumentando por lo tanto el número de países que requieren la aplicación de intervenciones específicas para educar a poblaciones más grandes, diagnosticar los trastornos, y asesorar a los pacientes afectados teniendo en cuenta estas intervenciones en su presupuesto de salud.
Aunque el conocimiento epidemiológico de la distribución, la prevalencia, la genética, la diversidad, y la carga sanitaria de la α-talasemia y la β-talasemia es limitado y en gran medida obsoleto, las brechas son más pronunciadas en el caso de la α-talasemia. Esta relativa falta de una base sólida de pruebas es probable que contribuya a la baja prioridad de este trastorno en muchas agendas de salud pública. Las revisiones existentes se centran principalmente en los aspectos moleculares y clínicos de la α-talasemia.
El objetivo de este artículo es proporcionar un resumen contemporáneo del conocimiento epidemiológico sobre la α-talasemia y discutir los diversos retos que enfrentan las comunidades médicas y de salud pública a la luz de los recientes descubrimientos sobre la gravedad y la genética de este trastorno heredado.
Relevancia Clínica
La hemoglobina adulta normal consiste en pares de cadenas α y β (α2β2), y la hemoglobina fetal tiene dos cadenas α y dos cadenas γ (α2γ2). Los genes para las cadenas α y las cadenas γ están duplicados (αα/αα, γγ/γγ), mientras que las cadenas β son codificadas por un locus único (β/β).
En el feto, la producción defectuosa de las cadenas α se refleja por la presencia de cadenas γ en exceso, que forman un tetrámero γ4, llamado hemoglobina de Bart; en los adultos, el exceso de cadenas β forman un tetrámero β4, llamado hemoglobina H (HbH). Debido a su muy alta afinidad por el oxígeno, ambos tetrámeros no pueden transportar oxígeno, y, en el caso de la HbH, su inestabilidad conduce a la producción de cuerpos de inclusión en los glóbulos rojos y a un grado variable de anemia hemolítica.
Hasta el momento se han identificado más de 100 formas genéticas de α-talasemia, con fenotipos que varían de asintomáticos a letales. A pesar de esta complejidad, la gravedad de este trastorno por lo general se correlaciona bien con el número de copias no funcionales de los genes de α-globina.
En base al número de genes de α-globina perdidos por deleción o inactivados total o parcialmente por mutaciones puntuales, las α-talasemias se clasifican en dos subgrupos principales: α+-talasemia (anteriormente llamada α-talasemia 2), en la que un par de los genes es eliminado o inactivado por una mutación puntual (-α/αα o ααND/αα, con ND denotando no deleción), y α0-talasemia (anteriormente llamada α-talasemia 1), en la que ambos pares de genes α-globina en el mismo cromosoma son suprimidos (--/αα).
Las formas clínicamente relevantes de α-talasemia por lo general implican a la α0-talasemia, ya sea co-heredada con α+-talasemia (-α/-- o ααND/--) resultando en enfermedad de HbH o heredada de ambos padres y resultando en hidropesía fetal por hemoglobina de Bart (--/--), que es letal en el útero o poco después del nacimiento. Los embriones afectados sucumben a la hipoxia severa ya sea temprano en la gestación (por ejemplo, en el caso de --FIL/--FIL [donde FIL se refiere a una deleción que causa α0-talasemia y que es prevalente entre los filipinos]) o durante el tercer trimestre (por ejemplo, en el caso de --SEA/--SEA [donde SEA se refiere a una deleción que causa α0-talasemia y que es frecuente entre personas del sudeste asiático]).
Pocos niños con hidropesía fetal por hemoglobina de Bart que recibieron una transfusión intrauterina o una transfusión inmediatamente después del parto han sobrevivido hasta los 5 años de edad. Estos niños requieren transfusiones periódicas y, cuando es apropiado, terapia de quelación del hierro; ellos por lo general tienen graves complicaciones clínicas, anomalías congénitas, y retrasos en las funciones cognitivas y motoras.
El síndrome de hidropesía fetal por hemoglobina de Bart a menudo se acompaña por una variedad de malformaciones congénitas y complicaciones maternas, incluyendo anemia severa del embarazo, preeclampsia, polihidramnios, y dificultades graves para expulsar el feto y la placenta sumamente agrandada. Aunque estas complicaciones han sido bien documentadas, hay datos muy limitados en cuanto a la frecuencia de las muertes maternas, sobre todo en los países en desarrollo en los que esta condición es tan común.
La enfermedad por HbH es considerada a menudo un trastorno relativamente leve. Sin embargo, los estudios han destacado ciertos fenotipos clínicamente graves, en particular en variantes no delecionales de la enfermedad. De hecho, la enfermedad por HbH se caracteriza por una amplia gama de características fenotípicas. La forma que resulta de deleciones (-α/--) por lo general sigue un curso relativamente leve, con anemia moderada y esplenomegalia.
Aparte de los episodios de infección intercurrente, esta forma de enfermedad por HbH no requiere transfusiones de sangre. Sin embargo, la variedad que resulta de la interacción de una mutación del gen α-globina no delecional junto con α0-talasemia (ααND/--) sigue un curso mucho más severo. Esto es particularmente cierto cuando la mutación no delecional es en la terminación de la cadena de α-globina de la hemoglobina mutante Constant Spring, que es muy común en muchos países asiáticos.
Las formas no delecionales de la enfermedad por HbH se caracterizan por una anemia grave, que ocurre a menudo en la vida temprana, y se asocian con esplenomegalia en aumento, una carga importante de hierro, y una variedad de otras complicaciones clínicas, incluyendo infecciones, úlceras en las piernas, cálculos biliares, y deficiencia de ácido fólico. Aunque en general está indicada la esplenectomía, la enfermedad por HbH no delecional se asocia con una tasa particularmente alta de complicaciones trombóticas. Esta observación hace que la decisión entre la esplenectomía y la transfusión de por vida sea extremadamente difícil.
Variantes más leves de α-talasemia actúan como modificadores genéticos de otras enfermedades hereditarias, como se ilustra por las interacciones epistáticas (cuando un gen influye en otro) entre la α-talasemia y la β-talasemia o entre la α-talasemia y la hemoglobina S (hemoglobina falciforme). Se han observado frecuentemente triplicaciones y cuadruplicaciones del gen de la α-globina en muchas poblaciones, y pueden interactuar con las variantes de β-talasemia para producir fenotipos más graves.
Por último, hay dos síndromes en los que la α-talasemia se asocia con retraso mental (Síndromes ATR). Los detalles relativos a estos síndromes se proporcionan en el Apéndice Suplementario, disponible con el texto completo de este artículo en NEJM.org.
Diagnóstico
Se requiere un diagnóstico prenatal para identificar a los fetos afectados por hidropesía fetal secundaria a hemoglobina de Bart y para reducir los riesgos para las madres. La decisión de considerar este diagnóstico por lo general lleva al hallazgo de glóbulos rojos microcíticos hipocrómicos en ambos padres, en asociación con un nivel normal de hemoglobina A2; esta combinación descartaría la β-talasemia, que por lo general implica un nivel elevado de hemoglobina A2. La deficiencia de hierro también tiene que ser descartada.
Cuando se dispone de instalaciones para el diagnóstico rápido de ADN, el examen hematológico es seguido por la confirmación de la presencia de α0-talasemia en los padres. El diagnóstico fetal por lo general se hace al principio del embarazo mediante una muestra de vellosidades coriónicas, aunque la anemia fetal también puede diagnosticarse después durante la gestación por cuantificación de la velocidad sistólica pico en la arteria cerebral media.
Varios métodos alternativos de diagnóstico genético pre-implantación y preconcepción o de diagnóstico prenatal - por ejemplo, análisis de ADN fetal en sangre materna e identificación de células fetales en sangre materna por tinción con anticuerpos contra las cadenas de globina - todavía están en etapas relativamente tempranas de estudio. Mientras tanto, los intentos de tratamiento intrauterino y postnatal se asocian con numerosos desafíos éticos.
El estado homocigoto de α+-talasemia y el estado heterocigoto de α0-talasemia (agrupados bajo el término "α-talasemia menor") se asocian con una reducción sustancial del volumen corpuscular medio (VCM) y de la hemoglobina corpuscular media (HCM). En los heterocigotos para α+-talasemia, el VCM y la HCM en general están disminuidos, pero hay una pequeña superposición con los valores normales. Las formas más leves de α-talasemia se diagnostican a menudo como deficiencia de hierro, aunque la frecuencia exacta de este diagnóstico erróneo es desconocida. En última instancia, el diagnóstico de una variante particular de la α-talasemia puede confirmarse sólo a nivel del ADN.
En la era pre-genómica, la frecuencia de α-talasemia se evaluó en base a la presencia de hemoglobina de Bart en sangre de cordón. La detección de hemoglobina de Bart en los recién nacidos indica que uno o más de los cuatro genes de α-globina son disfuncionales, causando α-talasemia. Aunque se pensó inicialmente que el nivel de hemoglobina de Bart al nacer sería un indicador sensible de la presencia de α-talasemia y que se correlacionaría bien con su gravedad, los estudios basados en ADN posteriores mostraron que este método diagnóstico falla en detectar un número sustancial de casos heterocigotos de α+-talasemia y, por lo tanto subestima la frecuencia de α-talasemia.
Actualmente está bien establecido que el diagnóstico de α-talasemia en base a la hemoglobina de Bart por sí sola no es confiable y no permite la identificación de los genotipos. Este método, sin embargo, sigue siendo ampliamente utilizado en países de bajos y medianos ingresos, porque es relativamente sencillo y mucho más barato que el análisis del ADN.
Distribución geográfica
La evidencia de que la α-talasemia es altamente protectora contra la malaria severa está bien establecida. Como resultado de esta ventaja selectiva, la α-talasemia heterocigota ha alcanzado altas frecuencias en todas las regiones tropicales y subtropicales, incluyendo la mayor parte del sudeste de Asia, el área del Mediterráneo, el subcontinente Indio, Oriente Medio y África.
Variantes comunes de α0-talasemia, predominantemente la mutación --SEA en el sudeste de Asia y la mutación --MED en el Mediterráneo, han llegado a frecuencias de aproximadamente 5%. Aunque hay por lo menos siete formas delecionales de α+-talasemia, las variantes -α3.7 son las más comunes. Se han reportado frecuencias del 70% y de hasta el 90% en Melanesia y en partes de Nepal, respectivamente.
Los mecanismos por los que se han alcanzado tales frecuencias cercanas a la fijación requieren mayor investigación. Además de los estudios que revelaron epistaxis negativa entre los pacientes con α+-talasemia y rasgo de células falciformes, resultando en un nivel reducido de protección contra la malaria cuando los dos son coheredados, modelos matemáticos han sugerido que la frecuencia de α+-talasemia podría estar limitada por la presencia del gen falciforme en África y el Mediterráneo.
En conjunción con los movimientos poblacionales globales a gran escala en las últimas décadas, la α-talasemia se ha extendido a muchas otras partes del mundo, incluyendo el norte de Europa y el Norte de América. Este fenómeno está mejor ilustrado por la aplicación en 1998 de un programa de cribado universal para α-talasemia en California. Después de la inmigración de un gran número de personas de Filipinas y de otros países del sudeste asiático, la incidencia de síndromes de α-talasemia en California entre enero de1998 y junio de 2006 fue de 11,1 casos por cada 100.000 personas evaluadas, con 406 casos de enfermedad por HbH y 5 casos de hidropesía fetal por hemoglobina de Bart.
Debido a la alta frecuencia de variantes de α+-talasemia en todo el mundo, es probable que, con la mezcla de poblaciones locales e inmigrantes, tal propagación aumente la incidencia de enfermedad por HbH, además de crear una carga de salud en un número creciente de países o regiones.
Varias encuestas de población sobre hemoglobinopatías revelaron heterogeneidades geográficas notables en la prevalencia de estos desórdenes. A mediados de la década de 1980, Flint y colegas observaron frecuencias de α+-talasemia que variaban del 6 al 68% a lo largo de las islas melanesias. Se describieron heterogeneidades similares más tarde en Vanuatu.
En una encuesta de micro-mapeo reciente llevada a cabo en Sri Lanka, la prevalencia de α-talasemia osciló entre el 2 y el 20% (Weatherall DJ, y col.: datos inéditos). Esta variabilidad es probable que sea el resultado de una serie de factores ambientales complejos, incluyendo la tasa de transmisión de la malaria. Una mejor comprensión de estas interacciones ayudará considerablemente en la refinación de las estimaciones de las poblaciones afectadas en las regiones tropicales y en las políticas de desarrollo para el diagnóstico y manejo de la α-talasemia en base a las características de la población local.
Es necesaria una cuantificación precisa de las poblaciones en riesgo de desarrollar síndromes de α-talasemia a nivel nacional, regional, y mundial para definir recursos actuales y futuros requeridos para proporcionar un diagnóstico prenatal preciso de variantes de α-talasemia, un manejo de emergencia de los embarazos con hidropesía fetal por hemoglobina de Bart y sus complicaciones maternas asociadas, y un manejo a largo plazo de la enfermedad por HbH.
Detección
Los objetivos principales de los programas de detección de talasemia son determinar la frecuencia de las diferentes variantes genéticas observadas en las comunidades e identificar e informar a las parejas que están en riesgo, en particular para las formas graves de la enfermedad presentes en las zonas de alta frecuencia.
La detección de la α-talasemia es especialmente útil en la prevención de complicaciones maternas graves en el caso de hidropesía fetal por hemoglobina de Bart, en la provisión de un diagnóstico preciso en los casos en los que la α-talasemia se cohereda con la hemoglobina S o la β-talasemia, o en casos en los que se detecta deficiencia de hierro. La mayoría de los encuestas de detección de α-talasemia son llevadas a cabo como parte de un programa de prevención de β-talasemia y por lo tanto no son adecuadas para determinar frecuencias de α-talasemia en la población.
Los beneficios del cribado de la población deberán considerarse cuidadosamente en cualquier población en la que las variantes de α-talasemia son frecuentes y cuando se observan casos de anemia microcítica hipocrómica inexplicable en ausencia de deficiencia de hierro.
Carga sanitaria y económica
Los primeros estudios sobre la carga global de los trastornos de la hemoglobina que evaluaron la frecuencia de los mismos y el número de años de vida ajustados por discapacidad (AVAD) asociado con ellos se vieron limitados por la falta de datos sobre la α-talasemia, en particular con respecto a la hidropesía fetal de la hemoglobina de Bart y a su carga en relación con los mortinatos o fallecidos poco después del parto.
La Organización Mundial de la Salud (OMS) no recopila datos sobre los mortinatos, y los únicos datos disponibles proveyeron una estimación de 1.250 embarazos con α0-talasemia homocigota por año en Tailandia, una cifra que corresponde a 37.242 AVAD. En comparación, se estimó que la β-talasemia homocigota y la hemoglobina E/β-talasemia resultaron en un total de 53.600 AVAD en Tailandia, en base a una esperanza de vida de 10 años y 30 años, respectivamente. Estas son probablemente subestimaciones porque se consideró la discapacidad de la madre sólo en el último trimestre del embarazo, sin tener en cuenta las complicaciones postnatales que pueden resultar en muerte, y porque no se calcularon las estimaciones para la enfermedad por HbH, ya que se realizó la recolección de los datos antes de que ocurriera una forma particularmente severa de la enfermedad por HbH en muchos países asiáticos.
La información sobre la incidencia de α-talasemia fue insuficiente para permitir el cálculo de la carga regional y global de enfermedad. Se han reportado recientemente estimaciones globales del número de AVAD resultantes de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad del 2010, aunque aún no se han añadido a este proyecto datos precisos sobre los diversos subtipos de talasemias.
Para el conocimiento de los autores, hay un solo estudio hasta la fecha que ha investigado la relación costo-beneficio de un programa de prevención de α-talasemia, establecido por sí mismo o como parte de un amplio programa para todas las talasemias o para condiciones genéticas (por ejemplo, fenilcetonuria) en general. El estudio, llevado a cabo en Hong Kong, concluyó que el cribado prenatal universal para α- y β-talasemias con el uso de pruebas de ADN era rentable, aunque no se dispuso de datos publicados sobre el costo del manejo de un embarazo afectado por hidropesía fetal secundaria a hemoglobina de Bart.
Debe investigarse cuidadosamente si estos resultados son aplicables a países con mayores poblaciones y menores ingresos que los de Hong Kong. Los beneficios del diagnóstico preciso de la deficiencia de hierro y otras hemoglobinopatías - Hemoglobina S y β-talasemia en particular - también deben ser considerados. Los análisis de costo-beneficio de los programas de prevención de β-talasemia en Quebec, Irán, Israel, y el Reino Unido generalmente han confirmado los beneficios generales de este tipo de programas (es decir, los costos de prevención fueron inferiores a los costos de tratamiento), pero los estudios no incluyeron a la α-talasemia.
En los países de bajos y medianos ingresos, esos programas podrían beneficiarse de las infraestructuras existentes (por ejemplo, los programas de cribado de fenilcetonuria o de deficiencia de glucosa-6-fosfato deshidrogenasa en curso) o podrían resultar en una mejora general del acceso a la atención de salud para las comunidades locales.
Manejo
El conocimiento detallado de la prevalencia de la α-talasemia (incluyendo el estado de portador) y de su diversidad genética es esencial para definir políticas orientadas a la reducción de la carga sanitaria a largo plazo de las hemoglobinopatías, permitiendo el diagnóstico preciso (y por lo tanto evitando investigaciones inexactas y caras), el establecimiento de la verdadera causa de la microcitosis, el asesoramiento genético adecuado, y la asignación de recursos para hacer frente a las emergencias y a las necesidades a largo plazo de los pacientes y sus familiares de una manera rentable. Para lograr estos objetivos, la comunidad médica se enfrenta a varios desafíos importantes, que se resumen en la Tabla 1.
Tabla 1. Principales desafíos asociados con la carga sanitaria en aumento de la α-talasemia y recomendaciones para superarlos
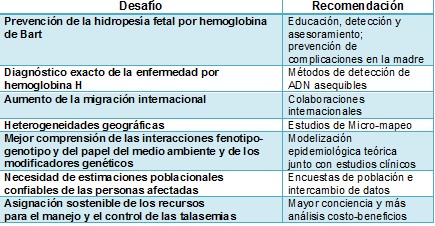 La mayoría de las parejas en riesgo de concebir fetos con hidropesía
fetal por hemoglobina de Bart no son actualmente identificadas. Por lo
tanto, es muy probable que las estimaciones actuales representen grandes
subestimaciones de los mortinatos por este desorden.
La mayoría de las parejas en riesgo de concebir fetos con hidropesía
fetal por hemoglobina de Bart no son actualmente identificadas. Por lo
tanto, es muy probable que las estimaciones actuales representen grandes
subestimaciones de los mortinatos por este desorden.
En ausencia de tratamientos específicos y de una clara comprensión de los mecanismos subyacentes responsables de la amplia gama de anomalías congénitas asociadas con este trastorno letal, el cribado y el diagnóstico prenatal temprano representan las únicas opciones para identificar los embarazos de alto riesgo y evitar complicaciones maternas graves.
Aunque por lo general se recomienda la interrupción de los embarazos afectados debido al aumento del riesgo de complicaciones maternas y fetales severas y a los efectos psicológicos en las familias, deben tenerse en cuenta los antecedentes culturales y religiosos cuando se asesora a las parejas en riesgo, tanto en las comunidades en las que la α-talasemia ha sido prevalente tradicionalmente como en aquellas en las que se ha introducido recientemente a través de la migración.
Un diagnóstico temprano correcto en el embarazo es esencial para evitar complicaciones médicas graves y trauma psicológico. Tal diagnóstico puede hacerse de forma confiable con costos relativamente bajos previendo que las mujeres embarazadas reciban un seguimiento regular por personal médico familiarizado con este síndrome.
Los recientes progresos en las pruebas de ADN revelaron una mayor diversidad fenotípica de la enfermedad por HbH que lo que se pensaba previamente.
Como se ha señalado anteriormente, las variantes no delecionales suelen ser más graves que las variantes de deleción. Por ello es crucial realizar el análisis del ADN para identificar la variante genética subyacente responsable de este trastorno. La reacción en cadena de la polimerasa-Gap (PCR) y los ensayos de PCR multiplex permiten una fácil detección de una gama de variantes comunes, pero tales métodos siguen siendo costosos y no son ampliamente utilizados en países de bajos y medianos ingresos. Con los métodos de prueba de ADN cada vez más y más asequibles, es probable que éstos se conviertan en una parte regular de los servicios disponibles para el control y manejo de las talasemias, en particular en los países asiáticos.
A medida que el conocimiento sobre la relación fenotipo-genotipo en la enfermedad por HbH mejora, la correcta identificación de genotipos será imprescindible para informar a los padres sobre los riesgos reproductivos durante el asesoramiento genético y para dar un adecuado cuidado a los pacientes afectados.
Con el aumento de los movimientos de población, la diversidad de combinaciones de variantes de α-talasemia o hemoglobinopatías coheredadas seguirá aumentando. Aunque es difícil predecir el fenotipo exacto de estas nuevas combinaciones, un conocimiento detallado de la distribución actual de las variantes genéticas ayudará al menos en la definición de los procedimientos de diagnóstico y en la evaluación de los riesgos potenciales.
Las colaboraciones entre las Áreas de "Origen" (aquellas con una alta prevalencia de α-talasemia y emigración sustancial) y las Áreas "Sumidero" (las que tienen inmigración sustancial de las zonas de origen), como las desarrolladas entre Filipinas y California y entre Sri Lanka y el Reino Unido, sin duda representan modelos beneficiosos para ambas partes y deben ser replicados más ampliamente.
La carga sanitaria de la α+-talasemia actualmente no se conoce. Aunque las variantes de α+-talasemia por si solas no representan un problema clínico directo, son probablemente los trastornos genéticos más comunes en el mundo, y representan importantes modificadores genéticos para una serie de condiciones, incluyendo la malaria, los trastornos de células falciformes, la β-talasemia, y la deficiencia de hierro.
Una mejor comprensión de estas interacciones es crucial para proporcionar un diagnóstico y tratamiento adecuado para los pacientes afectados, pero será relevante sólo si las intervenciones resultantes de esta mejor comprensión se implementan en todas las áreas en las que estas interacciones se están produciendo. Por ejemplo, se desconoce actualmente si las interacciones epistáticas entre el rasgo de células falciformes y la α+-talasemia observadas en Kenia también se están produciendo en las poblaciones indígenas.
Debido a la notable heterogeneidad geográfica en la prevalencia de la α-talasemia, las intervenciones tienen que adaptarse a las características específicas de la población local (por ejemplo, prevalencia del trastorno en la población, composición étnica, y consanguinidad) y al sistema de salud local. Una mejor comprensión de la relación entre la prevalencia de las variantes de α-talasemia, los factores ambientales y las infecciones, en combinación con métodos analíticos y de modelización modernos, ayudaría a lograr estimaciones más refinadas de las poblaciones afectadas. Tal conocimiento también ayudaría en el desarrollo de programas de prevención estratificados enfocados, por ejemplo, en regiones en las que las variantes de α0-talasemia son más frecuentes.
Conclusiones
Las α-talasemias representan un problema de salud mundial con una carga creciente. Un conocimiento refinado de las bases moleculares de la α-talasemia será totalmente relevante desde la perspectiva de la salud pública sólo si se complementa con datos epidemiológicos detallados.
Para garantizar el adecuado cuidado de los pacientes y la sostenibilidad de los sistemas de atención de salud, se debe poner mayor esfuerzo en obtener estimaciones basadas en la evidencia de las poblaciones afectadas, la provisión de recursos para la prevención, control y manejo de las talasemias, y realizar análisis de costo-efectividad. Tales objetivos se alcanzarán sólo a través de un esfuerzo concertado de las comunidades de investigación y médicas y el apoyo de agencias de financiación internacional para recopilar y compartir datos epidemiológicos. La reciente inclusión de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad y la evaluación de la carga de la α-talasemia en términos de AVAD tendrán sentido sólo si se dispone de datos confiables y actualizados.
Comentario:
Las talasemias son trastornos hereditarios de la síntesis de la hemoglobina que se caracterizan por una producción reducida de las cadenas de globina. Presentan una amplia distribución mundial debido al movimiento poblacional constante, pero no se cuenta actualmente con datos exactos de prevalencia. Estas migraciones de población contribuyen a la propagación de estas hemoglobinopatías aumentando el número de países que requieren la aplicación de intervenciones específicas para educar a sus comunidades, diagnosticar los trastornos, y asesorar a los pacientes afectados y a sus familias. Se requieren esfuerzos conjuntos para determinar las bases moleculares de las variantes de talasemia y su epidemiología a nivel global y regional, y para desarrollar programas de detección, manejo y asesoramiento con respecto a estos trastornos particulares de la hemoglobina.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
Introducción
| Las talasemias son las enfermedades humanas monogénicas más comunes. Estos trastornos hereditarios de la síntesis de la hemoglobina se caracterizan por una producción reducida de las cadenas de globina de la hemoglobina. |
Hay evidencia creciente de que la carga sanitaria y económica de las talasemias es cada vez mayor debido al crecimiento de la población, a la transición epidemiológica en regiones tropicales y a las migraciones humanas en otras partes del mundo. (La transición epidemiológica se refiere a un cambio en los patrones de distribución de las edades, la mortalidad, la fecundidad, la esperanza de vida, y las causas de muerte de la población, por lo general reflejado por un cambio de las muertes causadas por enfermedades infecciosas a las muertes causadas por enfermedades crónicas y degenerativas).
El crecimiento de la población conduce a un aumento absoluto del número de nacidos afectados. La transición epidemiológica mejora el diagnóstico de las hemoglobinopatías y la sobrevida de las personas afectadas, aumentando la incidencia de los trastornos. Las migraciones de la población, aunque no siempre conducen a un aumento de la prevalencia global, contribuyen a la propagación de las hemoglobinopatías aumentando por lo tanto el número de países que requieren la aplicación de intervenciones específicas para educar a poblaciones más grandes, diagnosticar los trastornos, y asesorar a los pacientes afectados teniendo en cuenta estas intervenciones en su presupuesto de salud.
Aunque el conocimiento epidemiológico de la distribución, la prevalencia, la genética, la diversidad, y la carga sanitaria de la α-talasemia y la β-talasemia es limitado y en gran medida obsoleto, las brechas son más pronunciadas en el caso de la α-talasemia. Esta relativa falta de una base sólida de pruebas es probable que contribuya a la baja prioridad de este trastorno en muchas agendas de salud pública. Las revisiones existentes se centran principalmente en los aspectos moleculares y clínicos de la α-talasemia.
El objetivo de este artículo es proporcionar un resumen contemporáneo del conocimiento epidemiológico sobre la α-talasemia y discutir los diversos retos que enfrentan las comunidades médicas y de salud pública a la luz de los recientes descubrimientos sobre la gravedad y la genética de este trastorno heredado.
Relevancia Clínica
La hemoglobina adulta normal consiste en pares de cadenas α y β (α2β2), y la hemoglobina fetal tiene dos cadenas α y dos cadenas γ (α2γ2). Los genes para las cadenas α y las cadenas γ están duplicados (αα/αα, γγ/γγ), mientras que las cadenas β son codificadas por un locus único (β/β).
En el feto, la producción defectuosa de las cadenas α se refleja por la presencia de cadenas γ en exceso, que forman un tetrámero γ4, llamado hemoglobina de Bart; en los adultos, el exceso de cadenas β forman un tetrámero β4, llamado hemoglobina H (HbH). Debido a su muy alta afinidad por el oxígeno, ambos tetrámeros no pueden transportar oxígeno, y, en el caso de la HbH, su inestabilidad conduce a la producción de cuerpos de inclusión en los glóbulos rojos y a un grado variable de anemia hemolítica.
Hasta el momento se han identificado más de 100 formas genéticas de α-talasemia, con fenotipos que varían de asintomáticos a letales. A pesar de esta complejidad, la gravedad de este trastorno por lo general se correlaciona bien con el número de copias no funcionales de los genes de α-globina.
En base al número de genes de α-globina perdidos por deleción o inactivados total o parcialmente por mutaciones puntuales, las α-talasemias se clasifican en dos subgrupos principales: α+-talasemia (anteriormente llamada α-talasemia 2), en la que un par de los genes es eliminado o inactivado por una mutación puntual (-α/αα o ααND/αα, con ND denotando no deleción), y α0-talasemia (anteriormente llamada α-talasemia 1), en la que ambos pares de genes α-globina en el mismo cromosoma son suprimidos (--/αα).
Las formas clínicamente relevantes de α-talasemia por lo general implican a la α0-talasemia, ya sea co-heredada con α+-talasemia (-α/-- o ααND/--) resultando en enfermedad de HbH o heredada de ambos padres y resultando en hidropesía fetal por hemoglobina de Bart (--/--), que es letal en el útero o poco después del nacimiento. Los embriones afectados sucumben a la hipoxia severa ya sea temprano en la gestación (por ejemplo, en el caso de --FIL/--FIL [donde FIL se refiere a una deleción que causa α0-talasemia y que es prevalente entre los filipinos]) o durante el tercer trimestre (por ejemplo, en el caso de --SEA/--SEA [donde SEA se refiere a una deleción que causa α0-talasemia y que es frecuente entre personas del sudeste asiático]).
Pocos niños con hidropesía fetal por hemoglobina de Bart que recibieron una transfusión intrauterina o una transfusión inmediatamente después del parto han sobrevivido hasta los 5 años de edad. Estos niños requieren transfusiones periódicas y, cuando es apropiado, terapia de quelación del hierro; ellos por lo general tienen graves complicaciones clínicas, anomalías congénitas, y retrasos en las funciones cognitivas y motoras.
El síndrome de hidropesía fetal por hemoglobina de Bart a menudo se acompaña por una variedad de malformaciones congénitas y complicaciones maternas, incluyendo anemia severa del embarazo, preeclampsia, polihidramnios, y dificultades graves para expulsar el feto y la placenta sumamente agrandada. Aunque estas complicaciones han sido bien documentadas, hay datos muy limitados en cuanto a la frecuencia de las muertes maternas, sobre todo en los países en desarrollo en los que esta condición es tan común.
La enfermedad por HbH es considerada a menudo un trastorno relativamente leve. Sin embargo, los estudios han destacado ciertos fenotipos clínicamente graves, en particular en variantes no delecionales de la enfermedad. De hecho, la enfermedad por HbH se caracteriza por una amplia gama de características fenotípicas. La forma que resulta de deleciones (-α/--) por lo general sigue un curso relativamente leve, con anemia moderada y esplenomegalia.
Aparte de los episodios de infección intercurrente, esta forma de enfermedad por HbH no requiere transfusiones de sangre. Sin embargo, la variedad que resulta de la interacción de una mutación del gen α-globina no delecional junto con α0-talasemia (ααND/--) sigue un curso mucho más severo. Esto es particularmente cierto cuando la mutación no delecional es en la terminación de la cadena de α-globina de la hemoglobina mutante Constant Spring, que es muy común en muchos países asiáticos.
Las formas no delecionales de la enfermedad por HbH se caracterizan por una anemia grave, que ocurre a menudo en la vida temprana, y se asocian con esplenomegalia en aumento, una carga importante de hierro, y una variedad de otras complicaciones clínicas, incluyendo infecciones, úlceras en las piernas, cálculos biliares, y deficiencia de ácido fólico. Aunque en general está indicada la esplenectomía, la enfermedad por HbH no delecional se asocia con una tasa particularmente alta de complicaciones trombóticas. Esta observación hace que la decisión entre la esplenectomía y la transfusión de por vida sea extremadamente difícil.
Variantes más leves de α-talasemia actúan como modificadores genéticos de otras enfermedades hereditarias, como se ilustra por las interacciones epistáticas (cuando un gen influye en otro) entre la α-talasemia y la β-talasemia o entre la α-talasemia y la hemoglobina S (hemoglobina falciforme). Se han observado frecuentemente triplicaciones y cuadruplicaciones del gen de la α-globina en muchas poblaciones, y pueden interactuar con las variantes de β-talasemia para producir fenotipos más graves.
Por último, hay dos síndromes en los que la α-talasemia se asocia con retraso mental (Síndromes ATR). Los detalles relativos a estos síndromes se proporcionan en el Apéndice Suplementario, disponible con el texto completo de este artículo en NEJM.org.
Diagnóstico
Se requiere un diagnóstico prenatal para identificar a los fetos afectados por hidropesía fetal secundaria a hemoglobina de Bart y para reducir los riesgos para las madres. La decisión de considerar este diagnóstico por lo general lleva al hallazgo de glóbulos rojos microcíticos hipocrómicos en ambos padres, en asociación con un nivel normal de hemoglobina A2; esta combinación descartaría la β-talasemia, que por lo general implica un nivel elevado de hemoglobina A2. La deficiencia de hierro también tiene que ser descartada.
Cuando se dispone de instalaciones para el diagnóstico rápido de ADN, el examen hematológico es seguido por la confirmación de la presencia de α0-talasemia en los padres. El diagnóstico fetal por lo general se hace al principio del embarazo mediante una muestra de vellosidades coriónicas, aunque la anemia fetal también puede diagnosticarse después durante la gestación por cuantificación de la velocidad sistólica pico en la arteria cerebral media.
Varios métodos alternativos de diagnóstico genético pre-implantación y preconcepción o de diagnóstico prenatal - por ejemplo, análisis de ADN fetal en sangre materna e identificación de células fetales en sangre materna por tinción con anticuerpos contra las cadenas de globina - todavía están en etapas relativamente tempranas de estudio. Mientras tanto, los intentos de tratamiento intrauterino y postnatal se asocian con numerosos desafíos éticos.
El estado homocigoto de α+-talasemia y el estado heterocigoto de α0-talasemia (agrupados bajo el término "α-talasemia menor") se asocian con una reducción sustancial del volumen corpuscular medio (VCM) y de la hemoglobina corpuscular media (HCM). En los heterocigotos para α+-talasemia, el VCM y la HCM en general están disminuidos, pero hay una pequeña superposición con los valores normales. Las formas más leves de α-talasemia se diagnostican a menudo como deficiencia de hierro, aunque la frecuencia exacta de este diagnóstico erróneo es desconocida. En última instancia, el diagnóstico de una variante particular de la α-talasemia puede confirmarse sólo a nivel del ADN.
En la era pre-genómica, la frecuencia de α-talasemia se evaluó en base a la presencia de hemoglobina de Bart en sangre de cordón. La detección de hemoglobina de Bart en los recién nacidos indica que uno o más de los cuatro genes de α-globina son disfuncionales, causando α-talasemia. Aunque se pensó inicialmente que el nivel de hemoglobina de Bart al nacer sería un indicador sensible de la presencia de α-talasemia y que se correlacionaría bien con su gravedad, los estudios basados en ADN posteriores mostraron que este método diagnóstico falla en detectar un número sustancial de casos heterocigotos de α+-talasemia y, por lo tanto subestima la frecuencia de α-talasemia.
Actualmente está bien establecido que el diagnóstico de α-talasemia en base a la hemoglobina de Bart por sí sola no es confiable y no permite la identificación de los genotipos. Este método, sin embargo, sigue siendo ampliamente utilizado en países de bajos y medianos ingresos, porque es relativamente sencillo y mucho más barato que el análisis del ADN.
Distribución geográfica
La evidencia de que la α-talasemia es altamente protectora contra la malaria severa está bien establecida. Como resultado de esta ventaja selectiva, la α-talasemia heterocigota ha alcanzado altas frecuencias en todas las regiones tropicales y subtropicales, incluyendo la mayor parte del sudeste de Asia, el área del Mediterráneo, el subcontinente Indio, Oriente Medio y África.
Variantes comunes de α0-talasemia, predominantemente la mutación --SEA en el sudeste de Asia y la mutación --MED en el Mediterráneo, han llegado a frecuencias de aproximadamente 5%. Aunque hay por lo menos siete formas delecionales de α+-talasemia, las variantes -α3.7 son las más comunes. Se han reportado frecuencias del 70% y de hasta el 90% en Melanesia y en partes de Nepal, respectivamente.
Los mecanismos por los que se han alcanzado tales frecuencias cercanas a la fijación requieren mayor investigación. Además de los estudios que revelaron epistaxis negativa entre los pacientes con α+-talasemia y rasgo de células falciformes, resultando en un nivel reducido de protección contra la malaria cuando los dos son coheredados, modelos matemáticos han sugerido que la frecuencia de α+-talasemia podría estar limitada por la presencia del gen falciforme en África y el Mediterráneo.
En conjunción con los movimientos poblacionales globales a gran escala en las últimas décadas, la α-talasemia se ha extendido a muchas otras partes del mundo, incluyendo el norte de Europa y el Norte de América. Este fenómeno está mejor ilustrado por la aplicación en 1998 de un programa de cribado universal para α-talasemia en California. Después de la inmigración de un gran número de personas de Filipinas y de otros países del sudeste asiático, la incidencia de síndromes de α-talasemia en California entre enero de1998 y junio de 2006 fue de 11,1 casos por cada 100.000 personas evaluadas, con 406 casos de enfermedad por HbH y 5 casos de hidropesía fetal por hemoglobina de Bart.
Debido a la alta frecuencia de variantes de α+-talasemia en todo el mundo, es probable que, con la mezcla de poblaciones locales e inmigrantes, tal propagación aumente la incidencia de enfermedad por HbH, además de crear una carga de salud en un número creciente de países o regiones.
Varias encuestas de población sobre hemoglobinopatías revelaron heterogeneidades geográficas notables en la prevalencia de estos desórdenes. A mediados de la década de 1980, Flint y colegas observaron frecuencias de α+-talasemia que variaban del 6 al 68% a lo largo de las islas melanesias. Se describieron heterogeneidades similares más tarde en Vanuatu.
En una encuesta de micro-mapeo reciente llevada a cabo en Sri Lanka, la prevalencia de α-talasemia osciló entre el 2 y el 20% (Weatherall DJ, y col.: datos inéditos). Esta variabilidad es probable que sea el resultado de una serie de factores ambientales complejos, incluyendo la tasa de transmisión de la malaria. Una mejor comprensión de estas interacciones ayudará considerablemente en la refinación de las estimaciones de las poblaciones afectadas en las regiones tropicales y en las políticas de desarrollo para el diagnóstico y manejo de la α-talasemia en base a las características de la población local.
Es necesaria una cuantificación precisa de las poblaciones en riesgo de desarrollar síndromes de α-talasemia a nivel nacional, regional, y mundial para definir recursos actuales y futuros requeridos para proporcionar un diagnóstico prenatal preciso de variantes de α-talasemia, un manejo de emergencia de los embarazos con hidropesía fetal por hemoglobina de Bart y sus complicaciones maternas asociadas, y un manejo a largo plazo de la enfermedad por HbH.
Detección
Los objetivos principales de los programas de detección de talasemia son determinar la frecuencia de las diferentes variantes genéticas observadas en las comunidades e identificar e informar a las parejas que están en riesgo, en particular para las formas graves de la enfermedad presentes en las zonas de alta frecuencia.
La detección de la α-talasemia es especialmente útil en la prevención de complicaciones maternas graves en el caso de hidropesía fetal por hemoglobina de Bart, en la provisión de un diagnóstico preciso en los casos en los que la α-talasemia se cohereda con la hemoglobina S o la β-talasemia, o en casos en los que se detecta deficiencia de hierro. La mayoría de los encuestas de detección de α-talasemia son llevadas a cabo como parte de un programa de prevención de β-talasemia y por lo tanto no son adecuadas para determinar frecuencias de α-talasemia en la población.
Los beneficios del cribado de la población deberán considerarse cuidadosamente en cualquier población en la que las variantes de α-talasemia son frecuentes y cuando se observan casos de anemia microcítica hipocrómica inexplicable en ausencia de deficiencia de hierro.
Carga sanitaria y económica
Los primeros estudios sobre la carga global de los trastornos de la hemoglobina que evaluaron la frecuencia de los mismos y el número de años de vida ajustados por discapacidad (AVAD) asociado con ellos se vieron limitados por la falta de datos sobre la α-talasemia, en particular con respecto a la hidropesía fetal de la hemoglobina de Bart y a su carga en relación con los mortinatos o fallecidos poco después del parto.
La Organización Mundial de la Salud (OMS) no recopila datos sobre los mortinatos, y los únicos datos disponibles proveyeron una estimación de 1.250 embarazos con α0-talasemia homocigota por año en Tailandia, una cifra que corresponde a 37.242 AVAD. En comparación, se estimó que la β-talasemia homocigota y la hemoglobina E/β-talasemia resultaron en un total de 53.600 AVAD en Tailandia, en base a una esperanza de vida de 10 años y 30 años, respectivamente. Estas son probablemente subestimaciones porque se consideró la discapacidad de la madre sólo en el último trimestre del embarazo, sin tener en cuenta las complicaciones postnatales que pueden resultar en muerte, y porque no se calcularon las estimaciones para la enfermedad por HbH, ya que se realizó la recolección de los datos antes de que ocurriera una forma particularmente severa de la enfermedad por HbH en muchos países asiáticos.
La información sobre la incidencia de α-talasemia fue insuficiente para permitir el cálculo de la carga regional y global de enfermedad. Se han reportado recientemente estimaciones globales del número de AVAD resultantes de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad del 2010, aunque aún no se han añadido a este proyecto datos precisos sobre los diversos subtipos de talasemias.
Para el conocimiento de los autores, hay un solo estudio hasta la fecha que ha investigado la relación costo-beneficio de un programa de prevención de α-talasemia, establecido por sí mismo o como parte de un amplio programa para todas las talasemias o para condiciones genéticas (por ejemplo, fenilcetonuria) en general. El estudio, llevado a cabo en Hong Kong, concluyó que el cribado prenatal universal para α- y β-talasemias con el uso de pruebas de ADN era rentable, aunque no se dispuso de datos publicados sobre el costo del manejo de un embarazo afectado por hidropesía fetal secundaria a hemoglobina de Bart.
Debe investigarse cuidadosamente si estos resultados son aplicables a países con mayores poblaciones y menores ingresos que los de Hong Kong. Los beneficios del diagnóstico preciso de la deficiencia de hierro y otras hemoglobinopatías - Hemoglobina S y β-talasemia en particular - también deben ser considerados. Los análisis de costo-beneficio de los programas de prevención de β-talasemia en Quebec, Irán, Israel, y el Reino Unido generalmente han confirmado los beneficios generales de este tipo de programas (es decir, los costos de prevención fueron inferiores a los costos de tratamiento), pero los estudios no incluyeron a la α-talasemia.
En los países de bajos y medianos ingresos, esos programas podrían beneficiarse de las infraestructuras existentes (por ejemplo, los programas de cribado de fenilcetonuria o de deficiencia de glucosa-6-fosfato deshidrogenasa en curso) o podrían resultar en una mejora general del acceso a la atención de salud para las comunidades locales.
Manejo
El conocimiento detallado de la prevalencia de la α-talasemia (incluyendo el estado de portador) y de su diversidad genética es esencial para definir políticas orientadas a la reducción de la carga sanitaria a largo plazo de las hemoglobinopatías, permitiendo el diagnóstico preciso (y por lo tanto evitando investigaciones inexactas y caras), el establecimiento de la verdadera causa de la microcitosis, el asesoramiento genético adecuado, y la asignación de recursos para hacer frente a las emergencias y a las necesidades a largo plazo de los pacientes y sus familiares de una manera rentable. Para lograr estos objetivos, la comunidad médica se enfrenta a varios desafíos importantes, que se resumen en la Tabla 1.
Tabla 1. Principales desafíos asociados con la carga sanitaria en aumento de la α-talasemia y recomendaciones para superarlos
En ausencia de tratamientos específicos y de una clara comprensión de los mecanismos subyacentes responsables de la amplia gama de anomalías congénitas asociadas con este trastorno letal, el cribado y el diagnóstico prenatal temprano representan las únicas opciones para identificar los embarazos de alto riesgo y evitar complicaciones maternas graves.
Aunque por lo general se recomienda la interrupción de los embarazos afectados debido al aumento del riesgo de complicaciones maternas y fetales severas y a los efectos psicológicos en las familias, deben tenerse en cuenta los antecedentes culturales y religiosos cuando se asesora a las parejas en riesgo, tanto en las comunidades en las que la α-talasemia ha sido prevalente tradicionalmente como en aquellas en las que se ha introducido recientemente a través de la migración.
Un diagnóstico temprano correcto en el embarazo es esencial para evitar complicaciones médicas graves y trauma psicológico. Tal diagnóstico puede hacerse de forma confiable con costos relativamente bajos previendo que las mujeres embarazadas reciban un seguimiento regular por personal médico familiarizado con este síndrome.
Los recientes progresos en las pruebas de ADN revelaron una mayor diversidad fenotípica de la enfermedad por HbH que lo que se pensaba previamente.
Como se ha señalado anteriormente, las variantes no delecionales suelen ser más graves que las variantes de deleción. Por ello es crucial realizar el análisis del ADN para identificar la variante genética subyacente responsable de este trastorno. La reacción en cadena de la polimerasa-Gap (PCR) y los ensayos de PCR multiplex permiten una fácil detección de una gama de variantes comunes, pero tales métodos siguen siendo costosos y no son ampliamente utilizados en países de bajos y medianos ingresos. Con los métodos de prueba de ADN cada vez más y más asequibles, es probable que éstos se conviertan en una parte regular de los servicios disponibles para el control y manejo de las talasemias, en particular en los países asiáticos.
A medida que el conocimiento sobre la relación fenotipo-genotipo en la enfermedad por HbH mejora, la correcta identificación de genotipos será imprescindible para informar a los padres sobre los riesgos reproductivos durante el asesoramiento genético y para dar un adecuado cuidado a los pacientes afectados.
Con el aumento de los movimientos de población, la diversidad de combinaciones de variantes de α-talasemia o hemoglobinopatías coheredadas seguirá aumentando. Aunque es difícil predecir el fenotipo exacto de estas nuevas combinaciones, un conocimiento detallado de la distribución actual de las variantes genéticas ayudará al menos en la definición de los procedimientos de diagnóstico y en la evaluación de los riesgos potenciales.
Las colaboraciones entre las Áreas de "Origen" (aquellas con una alta prevalencia de α-talasemia y emigración sustancial) y las Áreas "Sumidero" (las que tienen inmigración sustancial de las zonas de origen), como las desarrolladas entre Filipinas y California y entre Sri Lanka y el Reino Unido, sin duda representan modelos beneficiosos para ambas partes y deben ser replicados más ampliamente.
La carga sanitaria de la α+-talasemia actualmente no se conoce. Aunque las variantes de α+-talasemia por si solas no representan un problema clínico directo, son probablemente los trastornos genéticos más comunes en el mundo, y representan importantes modificadores genéticos para una serie de condiciones, incluyendo la malaria, los trastornos de células falciformes, la β-talasemia, y la deficiencia de hierro.
Una mejor comprensión de estas interacciones es crucial para proporcionar un diagnóstico y tratamiento adecuado para los pacientes afectados, pero será relevante sólo si las intervenciones resultantes de esta mejor comprensión se implementan en todas las áreas en las que estas interacciones se están produciendo. Por ejemplo, se desconoce actualmente si las interacciones epistáticas entre el rasgo de células falciformes y la α+-talasemia observadas en Kenia también se están produciendo en las poblaciones indígenas.
Debido a la notable heterogeneidad geográfica en la prevalencia de la α-talasemia, las intervenciones tienen que adaptarse a las características específicas de la población local (por ejemplo, prevalencia del trastorno en la población, composición étnica, y consanguinidad) y al sistema de salud local. Una mejor comprensión de la relación entre la prevalencia de las variantes de α-talasemia, los factores ambientales y las infecciones, en combinación con métodos analíticos y de modelización modernos, ayudaría a lograr estimaciones más refinadas de las poblaciones afectadas. Tal conocimiento también ayudaría en el desarrollo de programas de prevención estratificados enfocados, por ejemplo, en regiones en las que las variantes de α0-talasemia son más frecuentes.
Conclusiones
Las α-talasemias representan un problema de salud mundial con una carga creciente. Un conocimiento refinado de las bases moleculares de la α-talasemia será totalmente relevante desde la perspectiva de la salud pública sólo si se complementa con datos epidemiológicos detallados.
Para garantizar el adecuado cuidado de los pacientes y la sostenibilidad de los sistemas de atención de salud, se debe poner mayor esfuerzo en obtener estimaciones basadas en la evidencia de las poblaciones afectadas, la provisión de recursos para la prevención, control y manejo de las talasemias, y realizar análisis de costo-efectividad. Tales objetivos se alcanzarán sólo a través de un esfuerzo concertado de las comunidades de investigación y médicas y el apoyo de agencias de financiación internacional para recopilar y compartir datos epidemiológicos. La reciente inclusión de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad y la evaluación de la carga de la α-talasemia en términos de AVAD tendrán sentido sólo si se dispone de datos confiables y actualizados.
Comentario:
Las talasemias son trastornos hereditarios de la síntesis de la hemoglobina que se caracterizan por una producción reducida de las cadenas de globina. Presentan una amplia distribución mundial debido al movimiento poblacional constante, pero no se cuenta actualmente con datos exactos de prevalencia. Estas migraciones de población contribuyen a la propagación de estas hemoglobinopatías aumentando el número de países que requieren la aplicación de intervenciones específicas para educar a sus comunidades, diagnosticar los trastornos, y asesorar a los pacientes afectados y a sus familias. Se requieren esfuerzos conjuntos para determinar las bases moleculares de las variantes de talasemia y su epidemiología a nivel global y regional, y para desarrollar programas de detección, manejo y asesoramiento con respecto a estos trastornos particulares de la hemoglobina.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
No hay comentarios:
Publicar un comentario